PreDom チュートリアル
ドメイン間相互作用部位を調べたい
-
PreDom のサイドメニューで [PreDom:DiD]-[ドメイン間相互作用部位表示] を選択して PreDom:DiD の入力画面を表示します。
-
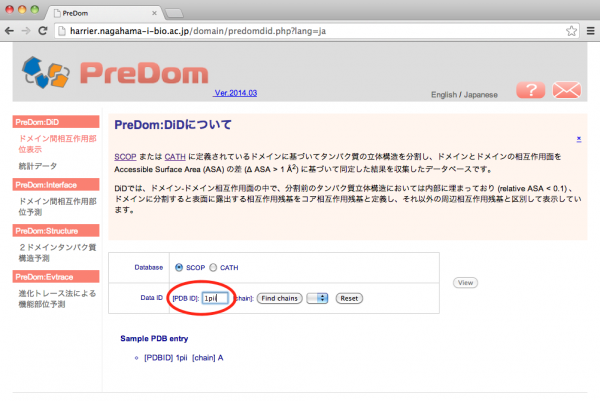
入力画面の [PDB ID] 欄に構造を見たいタンパク質の PDB ID を入力します。ここではサンプルデータの 1pii を使用します。
-
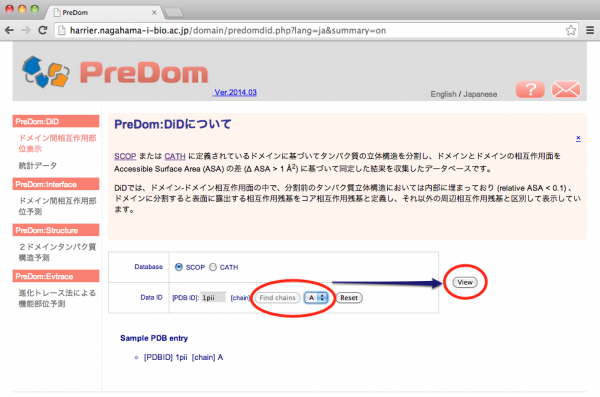
[Find chains] をクリックすると入力した PDB ID のタンパク質に含まれるchain が隣のドロップダウンリストに表示されるので、構造を見たいchain を選択します。必要に応じて [Database] でドメイン構造データベースを選択できますが、ここではデフォルト(SCOP)のままとします。[View] をクリックして検索を実行します。
-
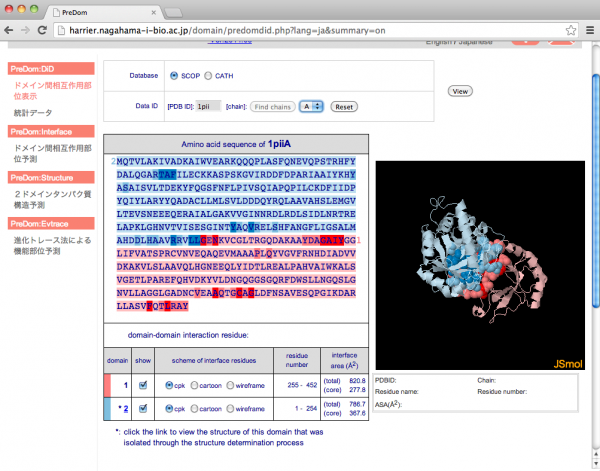
結果画面は以下の通りです。左側に指定された chain の配列及びドメイン情報、右側に JSmol による立体構造が表示されます。配列上で最も濃い色で表示されているのがコア相互作用残基、中間の色が周辺相互作用残基です。これらの相互作用残基は立体構造上では cpk で表示されています(初期状態)。
-
配列上の相互作用残基にマウスオーバーすると、その残基の色が白に切り替わって立体構造上の位置を確認することができます。また同時に残基名・残基番号が配列の下に表示されます。
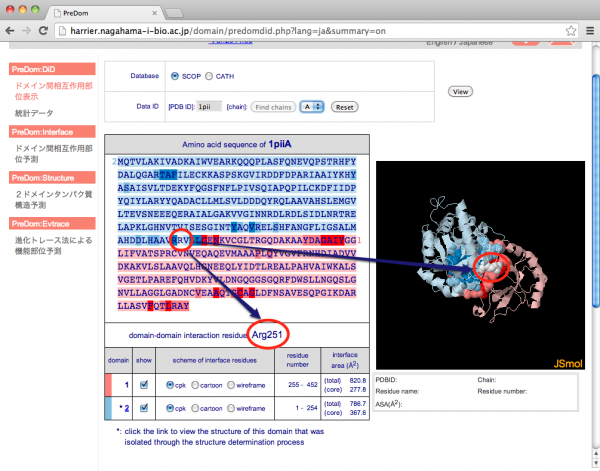
-
配列上の相互作用残基をクリックすると、立体構造の下の欄に残基の詳細情報が表示されます。
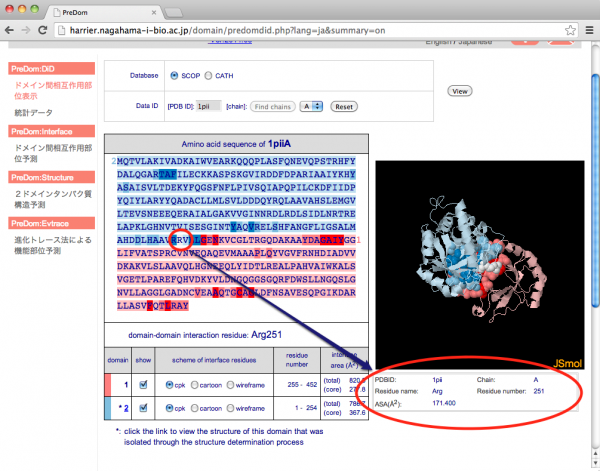
-
配列の下のドメイン情報欄のチェックボックスで各ドメインの表示/非表示を切り替えることができます。また、相互作用残基の表示形式はこの欄のラジオボタンで変更できます。
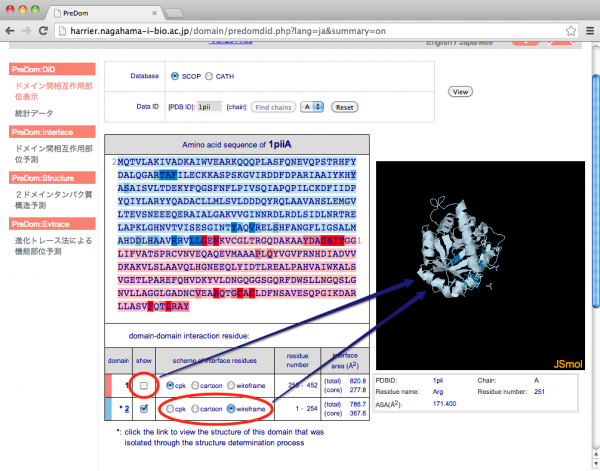
-
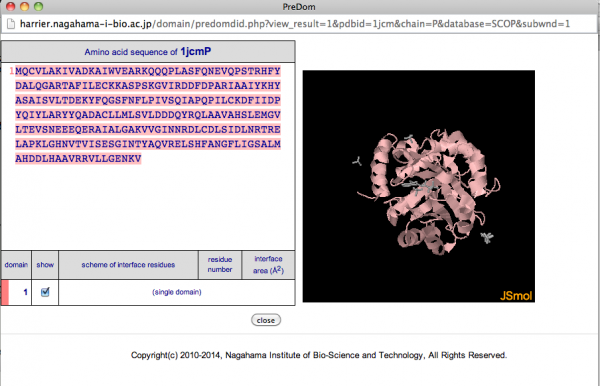
ドメイン番号にリンクが付加されているドメインは、配列類似性の高いシングルドメインタンパク質の構造が決定されており、単体で構造が決定されていると推定されるものです。リンクをクリックするとその配列及び立体構造が表示されます。下の図は 1piiA(ドメイン構造データベース:SCOP)の場合です。
[リンクが付加されたドメイン番号(1piiA の結果)]
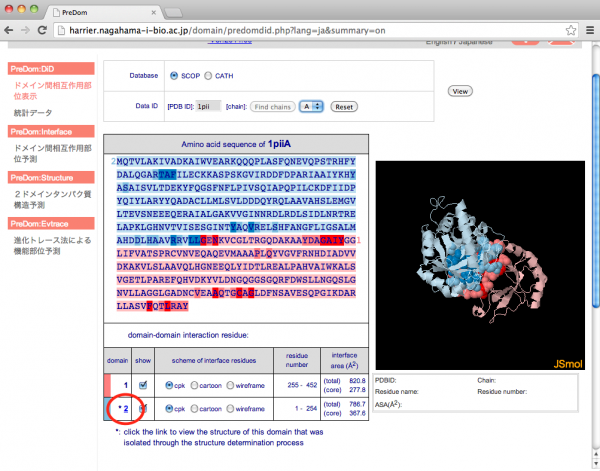
[単体での構造の推定結果]
-
PDB ID はそのままで chain またはドメイン構造データベースを変更して再検索する場合は、画面上部の入力欄でこれらを再選択して [View] をクリックします。

-
ドメイン間相互作用部位を予測したい
-
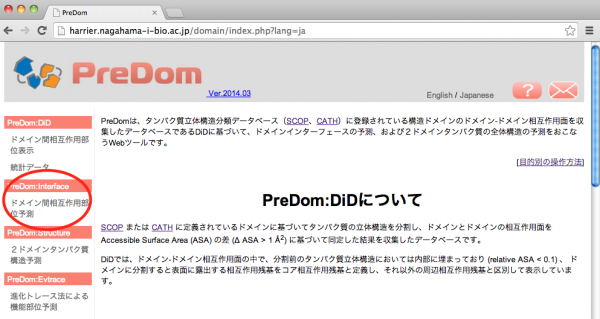
PreDom のサイドメニューで [PreDom:Interface]-[ドメイン間相互作用部位予測] を選択して PreDom:Interface の入力画面を表示します。
-
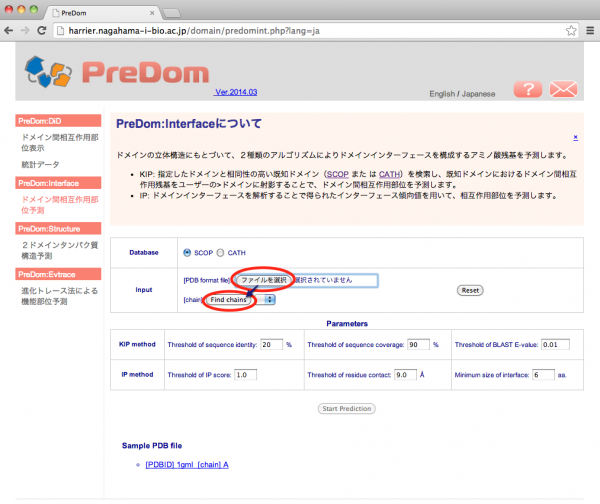
入力画面の [PDB format file] 欄で相互作用部位を予測したいタンパク質の PDB ファイルを選択します。ここではサンプルデータの 1gmlA.pdb を使用します。ファイル選択後 [Find chains] をクリックして選択した PDB データに含まれる chain を抽出します。
-
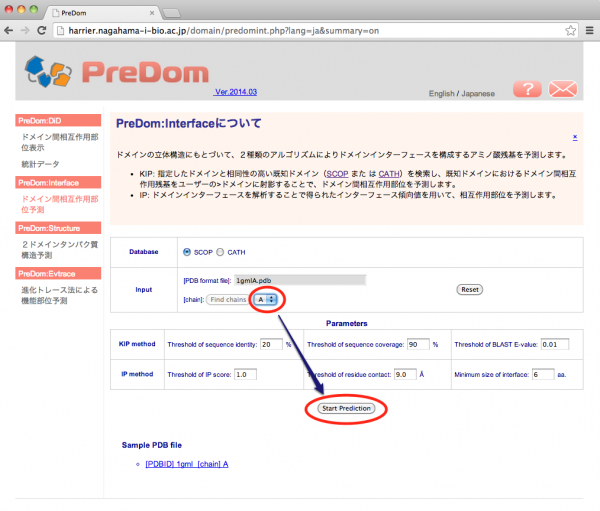
抽出された chain がドロップダウンリストに表示されるので、対象とする chain を選択します。ドメイン構造データベース及び予測アルゴリズムのパラメータは必要に応じて [Database] 及び [Parameters] 欄で変更できますが、ここでは全てデフォルトの値で実行します。[Start Prediction] をクリックして予測を実行します。
-
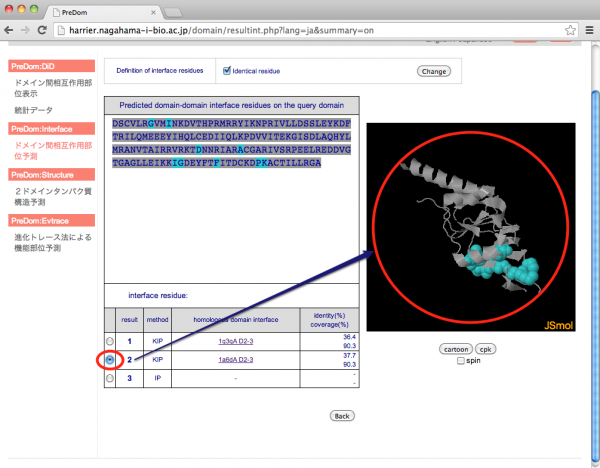
結果画面は以下の通りです。左側に指定された chain の配列及び予測結果リスト、右側に JSmol による立体構造が表示されます。青で表示されているのが予測された相互作用残基です。

-
初期状態では予測結果リストの一番上の結果が表示されています。他の結果を見る場合はリスト左端のラジオボタンで見たい結果を選択します。
-
配列上の相互作用残基にマウスオーバーすると、その残基の色が白に切り替わって立体構造上の位置を確認することができます。また同時に残基名・残基番号が配列の下に表示されます。

-
KIP の結果の [homologous domain] の ID に付加されているリンクをクリックすると、その相同ドメインの配列と入力配列のアラインメント及び相同ドメインの立体構造が表示されます。
[KIP の結果]

[相同ドメイン]

-
複数のドメインの立体構造からそれらの複合体構造を予測したい
-
PreDom のサイドメニューで [PreDom:Structure]-[マルチドメインタンパク質構造予測] を選択して PreDom:Structure の入力画面を表示します。

-
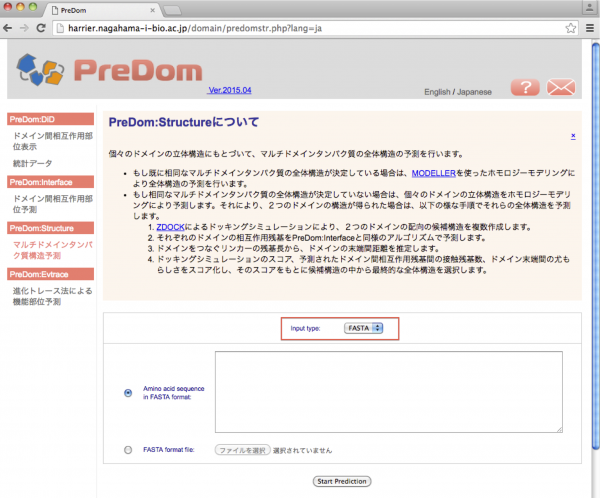
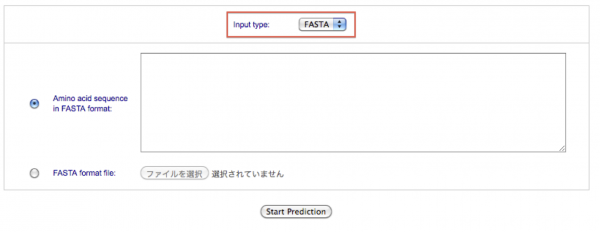
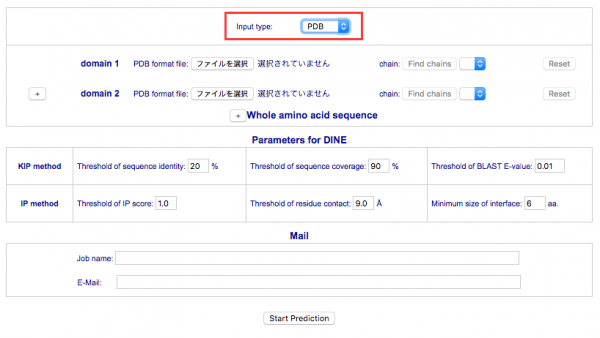
[Input type] のプルダウンメニューより、構造予測を行うタンパク質のデータ形式の種類を選択します。
- データ形式に FASTA を指定後の画面
- データ形式に PDB を指定後の画面
- データ形式に FASTA を指定後の画面
FASTA形式のアミノ酸配列を用いる場合
-
アミノ酸配列データを指定します。指定の方法は、立体構造を予測したいタンパク質のFASTA形式のアミノ酸配列データを、テキスト入力欄に直接入力する方法と、FASTA形式のアミノ酸配列データのファイルを指定する方法があります。
ここでは [Amino acid sequence in FASTA format:] を選択し、サンプルデータのFASTA形式のアミノ酸配列データを、テキスト入力欄にコピー&ペーストし、[Start Prediction] をクリックして予測を実行します。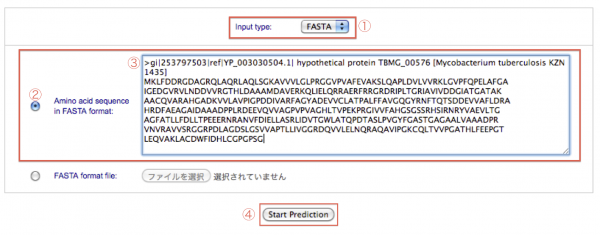
-
予測結果が表示されます。結果に応じて以下の4つの表示パターンがあります。
- 問い合わせ配列とほぼ同じ(配列一致度が95%以上の)立体構造既知タンパク質がある場合
- 問い合わせ配列と全長に渡って類似した(配列一致度が25%以上の)アミノ酸配列を持つ立体構造既知タンパク質がある場合
- 問い合わせ配列において、立体構造既知のドメイン、もしくはホモロジーモデリングにより立体構造予測可能なドメインが2つある場合
- 問い合わせ配列において、立体構造既知のドメイン、もしくはホモロジーモデリングにより立体構造予測可能なドメインが3つ以上ある場合
それぞれの場合の表示の詳細な説明は、ヘルプ画面あるいは「PreDom:Structure 使用マニュアル」を参照してください。
サンプルデータで予測を実行すると、立体構造予測可能なドメインが2つ得られます(ⅲのパターン)のでそれを例に取って説明します。問い合わせ配列において、立体構造既知のドメイン、もしくはホモロジーモデリングにより立体構造予測可能なドメインが2つある場合
- 立体構造予測可能なドメインが2つ得られた画面が表示されます。
- [Show] ボタン①をクリックすると、問い合わせ配列と鋳型タンパク質のアライメント、および鋳型タンパク質の立体構造が表示されます。立体構造既知のドメインの場合は、問い合わせ配列と立体構造既知ドメインとのアライメント、および立体構造既知ドメインの立体構造が表示されます。
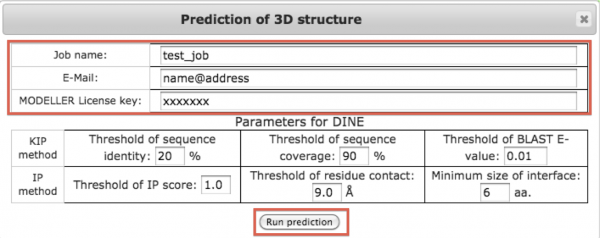
- [Status] の行にある [Click here] ②をクリックすると、DINEスコアによる全体構造予測を実行するウインドウが表示されます。このウインドウでは、Job名と予測完了通知メールの送信先、DINEスコアのパラメータを入力します。また、いずれかのドメインについてホモロジーモデリングにより立体構造を予測する場合は、MODELLERのライセンスキーの入力が必要です。DINEスコアのパラメータについては、ヘルプ画面の「PDB形式の立体構造データを用いる場合」の「パラメータの設定」を参照してください。
ここでは、立体構造予測を行いますので、MODELLERのライセンスキーを指定します。DINEスコアのパラメータはデフォルトのものを使用します。必要事項を入力し、[Run prediction] をクリックすると、ホモロジーモデリングおよびDINEスコアによる全体構造予測を開始します。 - 予測が終了すると、実行時に指定したメールアドレスにメールが送信されます。メールには予測された立体構造を表示するページへのURLが書かれています。
- 結果のページにアクセスすると、ホモロジーモデリングによりドメインを予測した場合は、予測されたドメインの構造をダウンロードすることができます。また、DINEスコアによる2つのドメインの全体構造予測結果も表示されます。なお、DINEスコアによる全体構造予測結果の見方については、ヘルプ画面の「DINEスコアによる2ドメインタンパク質の構造予測結果の見方」を参照してください。



[予測結果(画面上部)]
[予測結果(画面下部)]

PDB形式の立体構造データを用いる場合
-
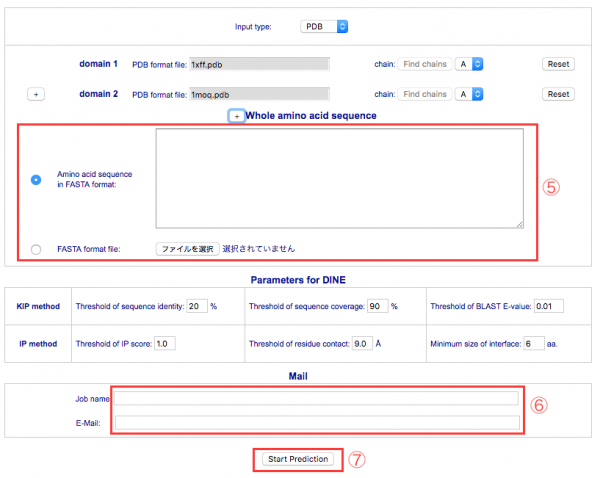
入力画面の [PDB format file] 欄の [ファイルを選択] ボタンをクリックし、元となるドメインの PDB データファイルを指定します①。3つめ以降のドメインのデータを入力するときは、 [+]ボタン③をクリックします。ドメインは10個まで指定できます。
注意 ファイルを指定する際は、必ずアミノ酸配列のN末側のドメインが上から順に並ぶようにPDBファイルを指定してください。順番通りに入力されていない場合、予測エラーとなります。
ここではサンプルデータの 1xffA と 1moqA を使用します。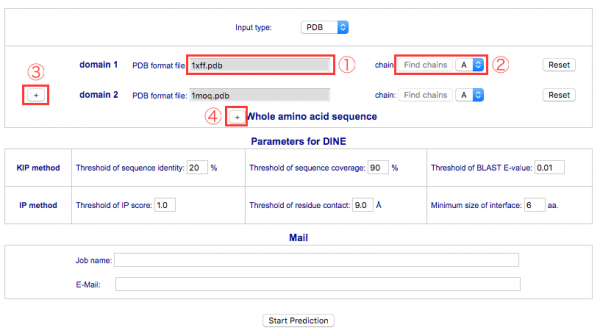
-
それぞれのドメインのファイルを指定後、隣りの [Find chains]② をクリックすると、指定したデータに含まれるチェーンのリストがその横のプルダウンメニューに生成されるので、その中から対象とするチェーンのIDを選択します。
-
全体構造のアミノ酸配列がわかっている場合は、[Whole amino acid sequence]の[+]ボタン④をクリックすると、アミノ酸配列データを入力するフィールド⑤が表示されます。配列の指定は、直接テキスト入力する方法とFASTA形式のファイルを指定する方法のいずれかを選択することができます。
[Whole amino acid sequence指定]
-
[Parameters for DINE] 欄では複合体構造予測に使用される相互作用部位予測アルゴリズムのパラメータを設定できます。各パラメータの詳細については、「PreDom:Structure」画面のヘルプ、または「PreDom:Structure 使用マニュアル」を参照してください。ここではデフォルトの値で実行します。
-
予測が終了すると、指定されたメールアドレスにメールを送信して通知します。[Job name] に実行するジョブを識別する任意の名称、[E-Mail] に通知を受け取るメールアドレスを入力し⑥、[Start Prediction]⑦ をクリックして予測を実行します。
-
予測開始を示す以下の画面が表示されます。この後十数分ほどで処理が完了し、「PreDom:Structure finished」という件名のメールで通知されます。
[予測開始画面]
[処理完了通知メールの文面]

-
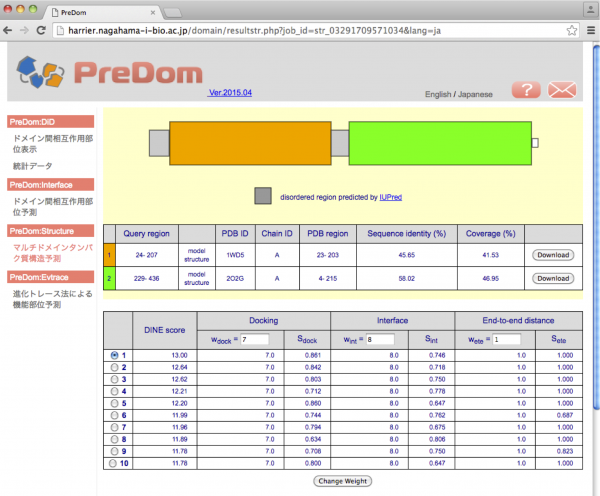
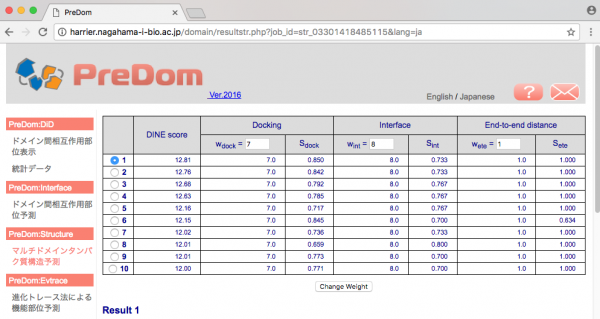
完了通知メールに記載されている [result page] の URL にアクセスすると予測結果が表示されます。画面上は候補構造リスト(スコアの高い順)、下は現在選択されている候補構造の配列及び立体構造です。
[予測結果(画面上部)]
[予測結果(画面下部)]

-
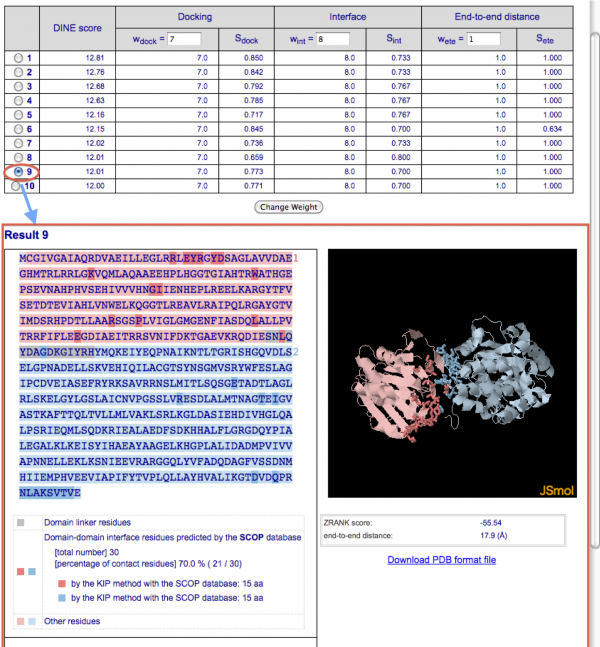
初期状態では候補構造リストの一番上の結果が表示されています。他の結果を見る場合はリスト左端のラジオボタンで見たい結果を選択します。
-
Docking, Interface, End-to-end distance の各スコアの重みはそれぞれテキストボックスで変更できます。入力後 [Change Weight] をクリックすると、DINE score が再計算され候補構造リストが更新されます。下の図では wdock を変更しています。
[スコアの重みの変更]

[変更した結果(wdock 変更)]

-
配列上の相互作用残基にマウスオーバーすると、その残基の色が白に切り替わって立体構造上の位置を確認することができます。また同時に残基名・残基番号が配列の下に表示されます。
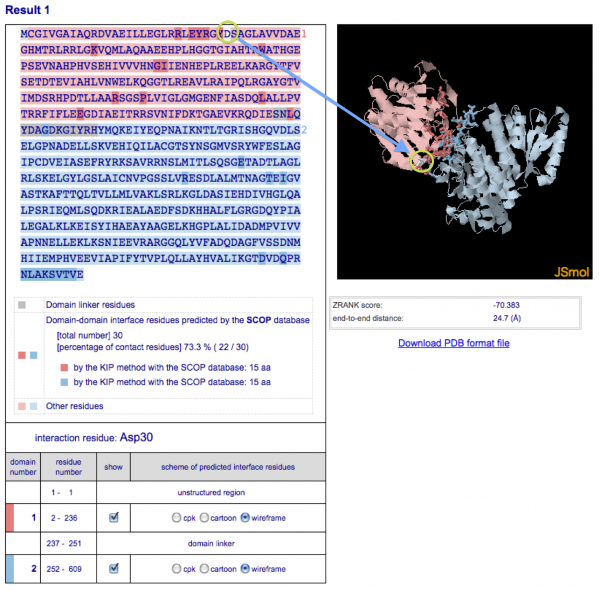
-
配列の下のドメイン情報欄のチェックボックスで各ドメインの表示/非表示を切り替えることができます。また、相互作用残基の表示形式はこの欄のラジオボタンで変更できます。
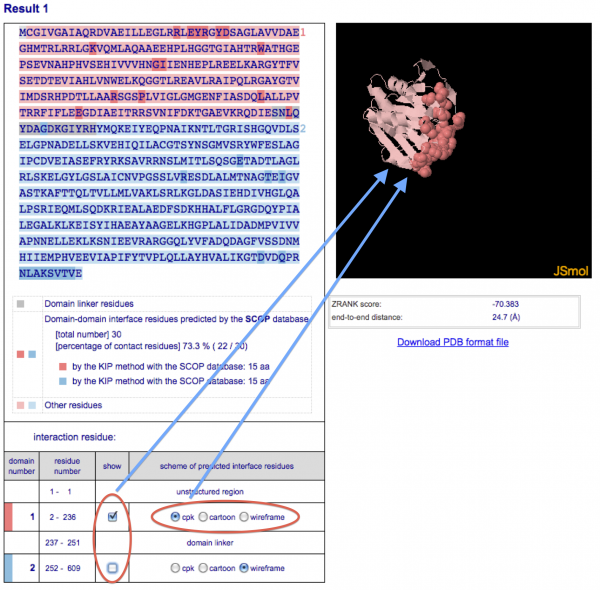
-
[Download PDB format file] のリンクをクリックすると、選択されている候補構造の PDB ファイルがダウンロードされます。

-
タンパク質の機能部位を予測したい
-
PreDom のサイドメニューで [PreDom:Evtrace]-[進化トレース法による機能部位予測] を選択して PreDom:Evtrace の入力画面を表示します。
-
[Input] のドロップダウンリストより入力データ形式(PDB ID + chain ID、PDB ファイル、FASTA ファイルのいずれか)を選択し、ID またはファイル名を入力します。ここでは PDB ID+chain ID のサンプル 1b6cB を使用します。[BLAST parameters] で必要に応じて BLAST のデータベース及び E-value 閾値を設定することができます。ここではデフォルト値のままで [BLAST] をクリックして次に進みます。

-
入力データの BLAST 検索でヒットした配列の一覧が表示されます。この中で選択された配列(赤で表示)が機能部位の予測に使用されます。初期状態では Identity ≥ 20(%), Coverage ≥ 90(%) を満たす配列が選択されていますが、Identity 及び Coverage をドロップダウンリストで変更、またはチェックボックスをチェック/チェック解除することにより変更できます(下の図は Identity を変更した状態)。ここでは初期状態のままで [Exec evtrace] をクリックして次に進みます。
[BLAST 検索結果]

[Identity の変更]
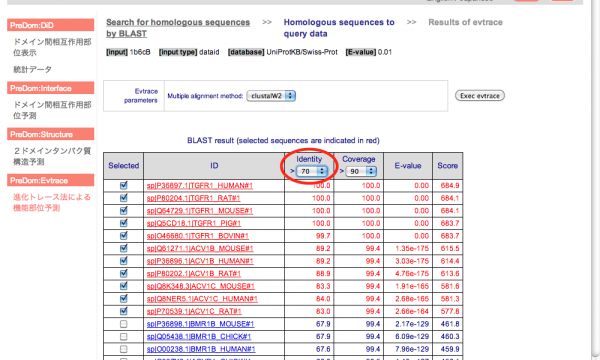
-
進化トレース法による機能部位の予測結果が表示されます。Tident をラジオボタンで変更すると下の表示が対応する結果に切り替わります。

-
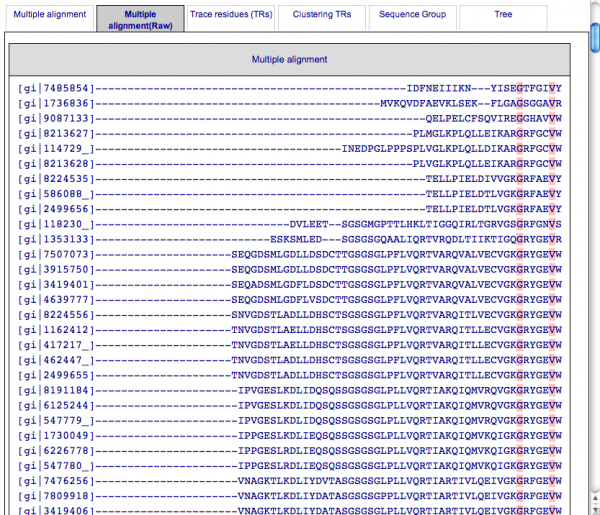
配列のマルチプルアラインメントを見るには画面中央部のタブ [Multiple alignment] もしくは [Multiple alignent (Raw)] を選択します。[Multiple alignment] のタブは1つのグループをグループ内で保存されたアミノ酸残基により表示しています。また、[Multiple alignment (Raw)] のタブは、前画面で選択したマルチプルアラインメントのプログラムにより得られたアラインメントそのものを表示しています。それぞれのタブではグループ間でも保存されている部位を赤で、グループで特異的に保存されている部位を灰色で表示しています。各グループの個々の配列を含む結果は [Download Multiple alignment result] をクリックするとダウンロードできます。
[[Multiple alignment] タブ]

[[Multiple alignment(Raw)] タブ]
[マルチプルアラインメントのダウンロード]

-
進化トレース法のトレース残基の分布を見るには画面中央部のタブ [Trace residues (TRs)] を選択します。配列上の conserved residue または class-specific residue にマウスオーバーすると、その残基の色が白に切り替わって立体構造上の位置を確認することができます。また同時に残基名・残基番号が配列の下に表示されます。

-
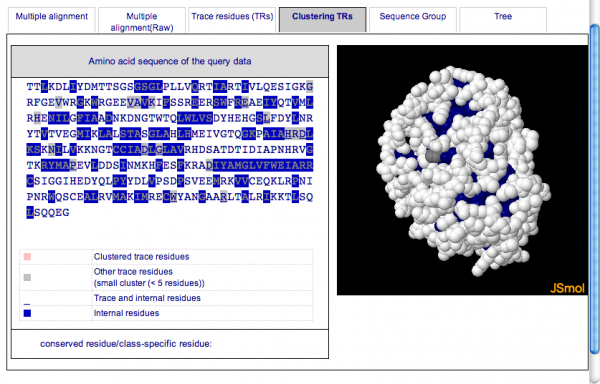
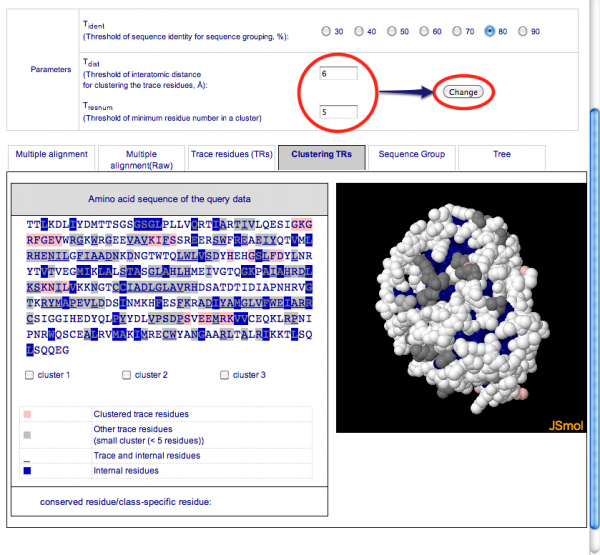
トレース残基のクラスタリングによって推定された機能部位を見るには画面中央部のタブ [Clustering TRs] を選択します。クラスターが形成されている場合は下の図(Tident = 80 の場合)のように配列の下にチェックボックスが表示され、チェックするとそのクラスターが配列及び立体構造上に色分けして表示されます。クラスタリングの条件を変更したい場合は上の [Parameters] で Tdist 及び Tresnum を変更して [Change] をクリックし、クラスタリングを再実行します。
[[Clustering TRs] タブ]
[クラスターの表示(Tident=80)/クラスタリングの再実行]
-
配列のグルーピングの結果を配列リストの形式で見るには画面中央部のタブ [Sequence Group] を選択します。配列がグループ毎に色分けされて表示されます。
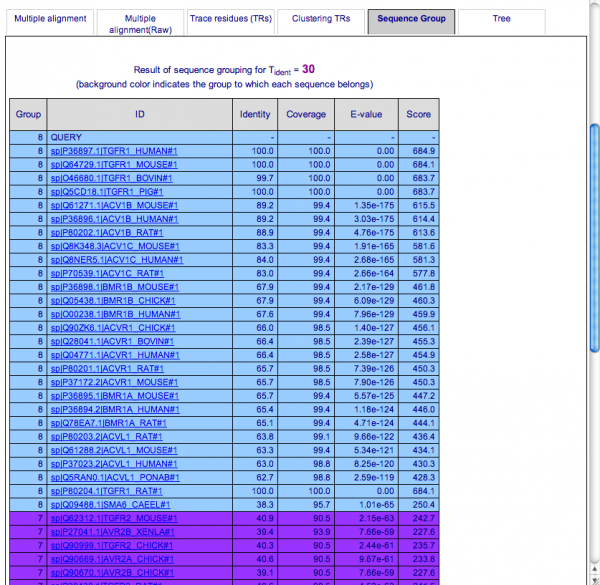
-
配列のグルーピングの結果を系統樹で見るには画面中央部のタブ [Tree] を選択します。
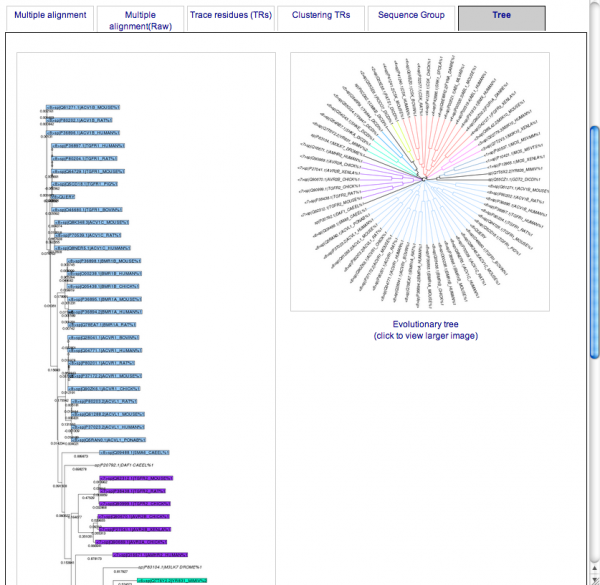
-